
Reaction coordinates for protein assembly and unfolding
Non-equilibrium dynamics of proteins, in particular their folding, unfolding, and assembly into supramolecular complexes, are vital determinants of both their function and pathology within living cells. Single molecule experimental techniques provide exquisite optical and mechanical means to probe and control the structure and non-equilibrium dynamics of proteins. However, such techniques lack high resolution atomistic level information on the response of molecules to such external controls. Computational methods are ideally suited to fill the void of missing information through detailed high resolution modelling of non-equilibrium dynamics or the response of biomolecules under external forces. Our program has made specific contributions in extracting the transient intermediates and effective reaction coordinates during both protein aggregation and unfolding, as described below.
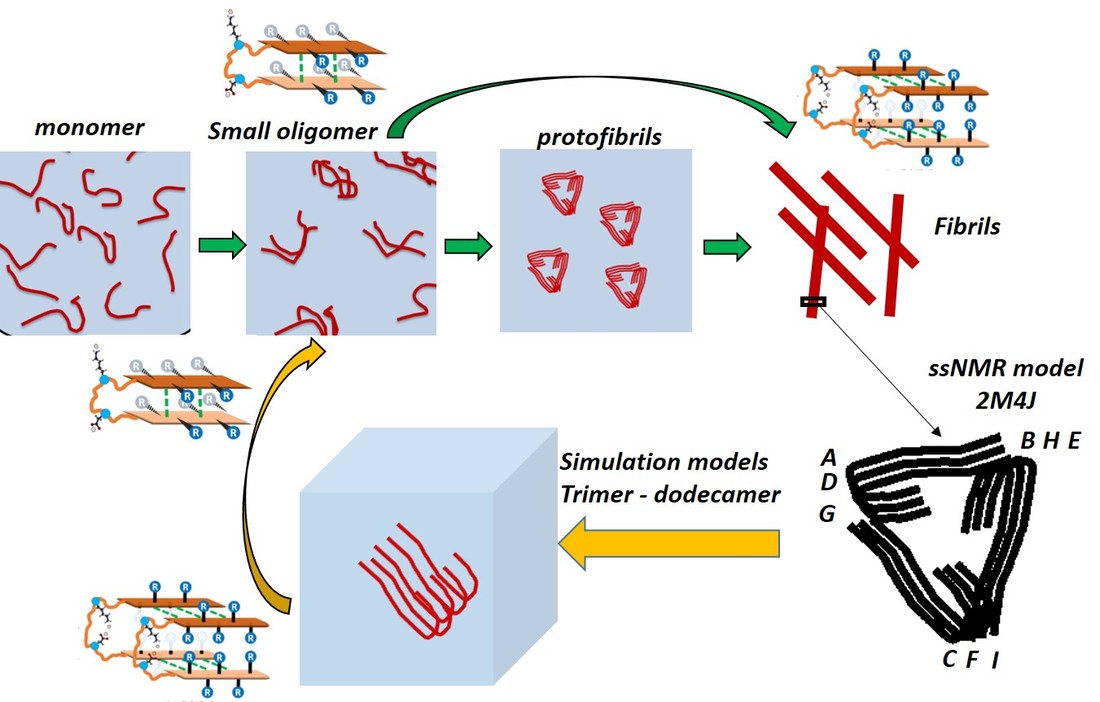
Reaction Coordinates for Molecular Association and AggregationAlzheimer’s disease (AD), a neurodegenerative disorder, is characterized by the presence of extracellular deposits of plaque and intracellular neurofibrillary tangles. The plaques form mostly due to aggregation followed by fibrillation of amyloid beta (Aβ) peptide. Recent studies have shown that although plaques are composed of fibrils, small oligomeric assemblies of the peptide are the major toxic species. However, the structural basis for this toxicity remains unclear as the oligomer species is transient and inaccessible to commonly used molecular structure determination techniques. Using innovative optical techniques, our experimental collaborators found signatures of hairpin conformations stabilized within an oligomeric assembly along the aggregation pathway. Transient, anti-parallel β-sheet hairpin structures had been reported in previous computational studies and had been trapped previously using antibodies or chemical modifications. However, there was no evidence that anti-parallel hairpin monomer conformations can be stabilized within small wild-type Aβ oligomers.
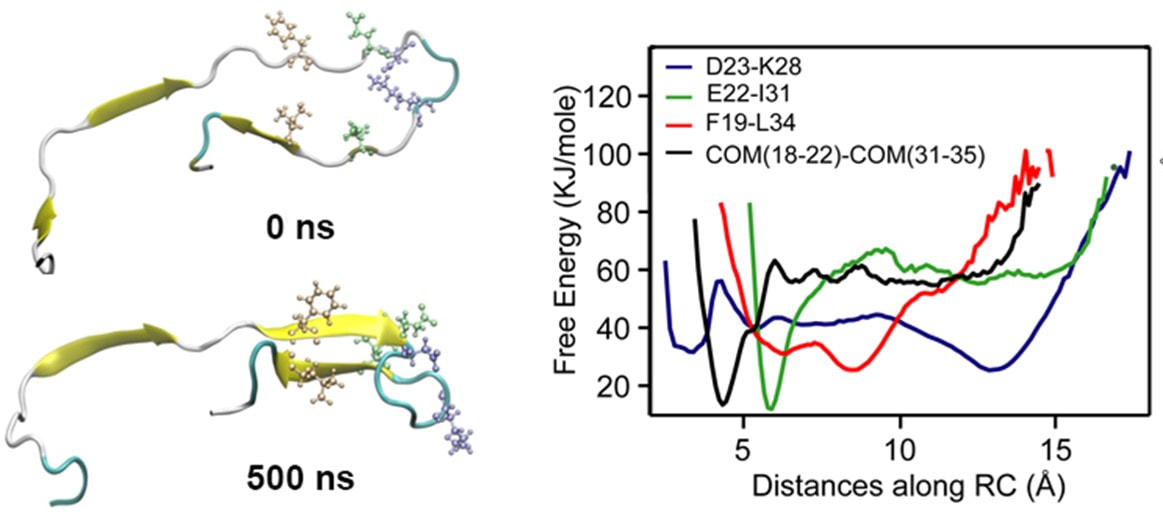
Using atomisitic simulations we examined oligomer structures of different sizes ranging from trimer to dodecamer in solution (Figure: top right), to capture and characterize an unbiased secondary structure transition associated with monomer units embedded within small oligomers and fibrils. The structural signatures (Figure: bottom left) captured during the fibril-like monomer to oligomer-like monomer state were: 1) formation of anti-parallel β-sheets, and 2) dissolution of the salt bridge between D23 and K28, both consistent with experimental signatures. Our simulations show for the first time that anti-parallel β-sheet hairpin peptide conformations can be stabilized within pentamer and hexamer Aβ assemblies over hundreds of nanoseconds. 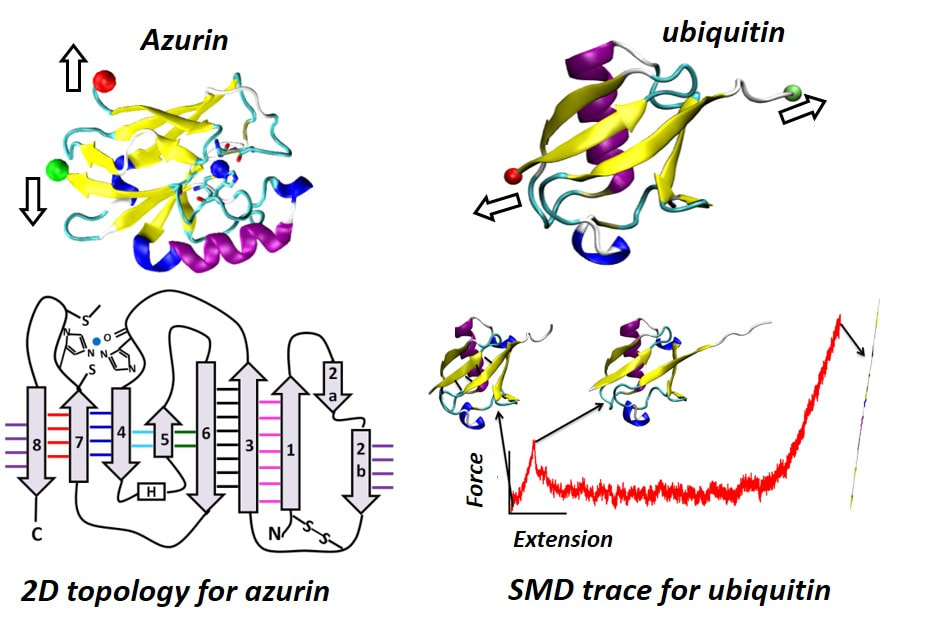
|
Mechanical Unfolding Pathways of ProteinsSingle molecule force spectroscopy (SMFS) experiments directly measure mechanical properties of proteins and protein complexes, such as the directional mechanical stability and stiffness. Our group has developed computational strategies for rigorous collective analysis of hundreds of atomistic steered molecular dynamics (SMD) simulation trajectories and applied it to capture transition states and reaction coordinates accessed during mechanical unfolding of proteins in SMFS experiments in the labs of our collaborators. We are studying two challenging systems (Figure on the left), viz. the metalloprotein azurin, and proteins from the ubiquitin family.
For azurin, we are nvestigating the effect of copper binding and the disulphide bond on the stability and unfolding pathways of the protein. Our studies have thus far been able to resolve subtle features in the unfolding force traces induced by copper binding. We have also been able to resolve stability differences in ubiqutin family proteins and identify specific secondary strucutre elements responsible for the differences. |
